Highlights
The molecular architecture of the authentic SARS-CoV-2 virus is unveiled by cryo-electron tomography and subtomogram averaging.
The native structures and landscape of the SARS-CoV-2 spikes revealed.
The native compositions of the glycans are characterized by mass spectrometry.
Structure and assembly of the SARS-CoV-2 viral RNPs are revealed in situ for the first time.
All structures are deposited in the Electron Microscopy Data Bank, providing molecular basis for drug and vaccine development, publicity of epidemic prevention, public education and molecular dynamic simulation.
The architecture of the intact SARS-CoV-2 virus is revealed for the first time
Introduction
In 2020, SARS-CoV-2, the virus that causes COVID-19, spread around the world. The pandemic attracted global attention to the enveloped viruses, which include the SARS-CoV-2 virus. The 2020 Nobel Prizes in Physiology or Medicine was awarded to the researchers who discovered the hepatitis C virus. It was a great encouragement for researchers who have persisted in the studies of enveloped viruses. However, there are still enormous misunderstandings of the public about infectious viruses. The SARS-CoV-2 virus that causes COVID-19 was once an “invisible enemy” such that people lacked awareness for the prevention of the pandemic.
As early as in January, Sai Li, a researcher from School of Life Science at Tsinghua University, made up his mind to post the high resolution “wanted poster” of the SARS-CoV-2 virus to the world. By combining cryo-electron tomography (cryo-ET) and subtomogram averaging (STA), a joint effort of Sai Li’s group and Lanjuan Li’s group from State Key Laboratory for Diagnosis and Treatment of Infectious Diseases at Zhejiang University has recently revealed the three-dimensional architecture of the intact, authentic SARS-CoV-2 virus. This study, titled “Molecular Architecture of the SARS-CoV-2 Virus”, has been published in Cell on September 15, 2020.

Molecular architecture of the authentic SARS-CoV-2 virus
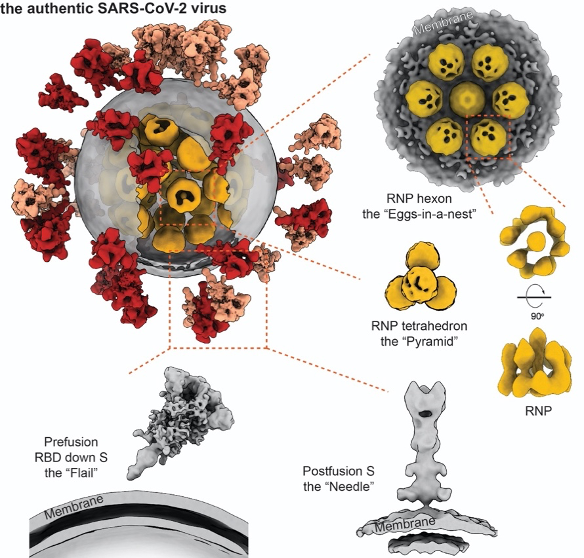
In this study, the architecture of the SARS-CoV-2 virus was revealed at molecular resolution, providing a structural overview on how the virus looks like. In this collaboration, the group at Zhejiang University provided the chemically inactivated virus sample, which preserves the virion in its most authentic structure. Researchers at Tsinghua University purified the virions in a biosafety-level 2 laboratory and subsequently plunge-froze them. Using the their locally developed high throughput, high resolution cryo-electron tomography (cryo-ET) workflow, the researchers collected over 100 TB tomographic data. From these 3D data, 2,294 intact virions (the largest cryo-ET data set about SARS-CoV-2 virus to date) were selected, and 55,000 spike (S) proteins, 20,000 ribonucleoproteins (RNPs) were further picked from these virions for subsequent structural analysis. Eventually, a representative virus was reconstructed, unveiling the molecular architecture of an intact SARS-CoV-2 at resolutions up to 7.8Å. The statistical analysis suggests that the average diameter of the virions’ envelope was about 80 nm; approximately 30 Spikes were distributed on each virion and about 30 RNPs were in its lumen.

A cryo-ET image of SARS-CoV-2 viruses with a computer reconstruction of one virus
Native structures of Spike
The coronavirus gained its name from its crown-like spikes, which protrude from the surface of the virus and act as a key for the virus’s invasion into the host cell. Therefore, most vaccines and antibodies are developed to target the spikes of SARS-CoV-2.
The study has resolved the structures of spikes in RBD down, one RND up, and postfusion conformations. It was observed that the SARS-CoV-2 spikes distribute randomly on the viral envelope, and the prefusion spikes can swing almost freely around their stems just like a “flail”. These unique features allow the spikes to explore the surrounding and better bind to the receptors on the vulnerable cells, further facilitate the virus’s invasion. On the other hand, the spikes are also found to be fragile. Improper methods of inactivation and purification may lead to the shedding of one subunit or even the whole spike, resulting in abnormal “bald” virions and low efficiency for developing vaccines to induce neutralizing antibodies.
The surface of spike is densely covered with 66 N-linked glycans, which serve as a “shield” to avoid host recognition and neutralization. There has been no detailed research about the compositions of the native spike glycosylation. Aided by the Proteinomics Facility at the Technology Center for Protein Sciences, the native glycan identity of the SARS-CoV-2 spike was analyzed by mass spectrometry (MS). It was found that the native glycans were larger and more complex than native glycans, however can still be termed as similar to the recombinant spike. Such similarity provided confidence for the effectiveness of protein-based vaccines against SARS-CoV-2.
Structure and assembly of the RNPs of positive-strand RNA virus are revealed for the first time
Coronaviruses have the largest genome among all RNA viruses, that the total length of genome RNA may reach 100 times as the virion’s diameter. All genetic information of the SARS-CoV-2 virus is encoded in its ribonucleic acid (RNA). Protecting the genome from damaging is a vital task for the virus. Arranging and packaging the long genomic strand into a tiny lumen while avoiding its entangling, knotting or even damaging, remains to be a geometric challenge for all viruses. This role is carried out by the the nucleocapsid (N) proteins. It assembles with the single-stranded RNA into a ribonucleoprotein (RNP), which are further packaged into the viral lumen. RNPs are also in charge of orderly releasing the RNA into infected cell plasma after membrane fusion. However, neither the structure or the packaging mechanism was known to the RNPs of all positive-strand RNA viruses, including the SARS-CoV-2 virus.
Researchers picked and analyzed nearly 20,000 RNPs in the lumen of the SARS-CoV-2 virus, and enlightened the architecture and assembly of RNPs for the first time. The native RNPs organize the single-stranded RNA in a “beads-on-a-string” stoichiometry, and are packed into the viral lumen as an “eggs in a nest”-shaped hexagonal assembly or a “pyramid”-shaped tetrahedral assembly. The densely assembled RNPs also strengthen the SARS-CoV-2 virus, enforcing it against the environmental and physical challenges. This was the first time to enlighten the lumen of positive-strand RNA viruses, which turns out to be very distinctive from the well-studied negative-strand RNA viruses.
Responses from academia and public
The reconstructed three-dimensional map of the SARS-CoV-2 virus allows this “invisible enemy” to be clearly displayed, shows people more detailed about the authentic SARS-CoV-2 structure. Professor Felix A. Rey, an esteemed structural biologist and virologist, and a member of the French Academy of Sciences, congratulated this achievement in a letter, “It is a milestone” he said.
The work has been repeatedly reported by major press media around the world. All structural data has been shared in the Electron Microscopy Data Bank and can be accessed for free, providing molecular basis for drug and vaccine development, publicity of epidemic prevention, public education, and molecular dynamic simulation.

Group photo of Sai Li’s group. From the left: Zheyuan Zhang, Yutong Song, Chujie Sun, Sai Li, Yong Chen, Jiaxing Zhang, Jialu Xu
Sai Li (Tsinghua University) is the lead contact of this article. Lanjuan Li (Zhejiang University) is the co-corresponding author. Hangping Yao (Zhejiang University), Yutong Song (Tsinghua University), Yong Chen (Tsinghua University), Nanping Wu (Zhejiang University), and Jialu Xu (Tsinghua University) are the co-first authors. In addition, Chujie Sun, Jiaxing Zhang, and Zheyuan Zhang from Sai Li's group also participated in the work. The project was supported by several experts in structural biology and virology, including Yigong Shi (Tsinghua University), Max Crispin (University of Southampton), and Jianlin Lei (Tsinghua University). We are in debt to Dr. Hongwei Wang, Dr. Nieng Yan, Dr. Xinquan Wang, Dr. Lingqi Zhang, Dr. Qiang Ding, and Dr. Xueming Li for critical advice.
Thanks to the Tsinghua University Branch of China National Center for Protein Sciences (Beijing) and the Proteinomics Facility at the Technology Center for Protein Sciences for facility supports. This work is supported by the start-up fund of Tsinghua University, Beijing Advanced Innovation Center for Structural Biology & Frontier Research Center for Biological Structure, and THU-PKU Center for Life Sciences.
Links:
https://www.cell.com/cell/fulltext/S0092-8674(20)31159-4
https://doi.org/10.1016/j.cell.2020.09.018